Protocols and tips in protein purification or How to purify protein in one day.
Among about of dozen kinds of chromatography the most universal and useful are:
| Cromatography type | Purification fold |
| Ion-exchange chromatography | 2 - 10 |
| Gel-filtration | 2 - 3 |
| Hydrophobic chromatography | 2 - 10 |
| Affinity and pseudo affinity chromatography | up to hundreds |
To become familiar with the basic theory and methods in chromatography, please read the useful booklets from Amersham Bioscience. You can get copies of them on the Web... From the list of handbooks you need to read at least 5:
- Affinity chromatography.
- Principles and Methods Ion exchange chromatography.
- Principles and Methods Gel filtration.
- Principles and Methods Hydrophobic interaction chromatography.
- Principles and Methods Protein Purification Handbook.
Scheme of the chromatographic system:
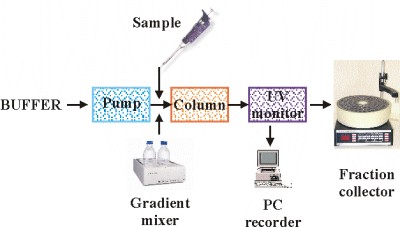
The simplest chromatographic system consists of a peristaltic pump, column and fraction collector. A gradient mixer is required if gradient elution of the proteins is used. Modern sophisticated systems have all the components built in and are operated by PC.
NB
Please notice that the pump is placed before the column, not after. Don't place the horse behind the cart!
Sequence of operations during ion exchange or hydrophobic chromatography:
- Connect chosen column to the system
- Wash with 1-2 column bed volumes (CBV) of starting buffer
- Apply sample. Collect unbound material in the separate container or start to collect fractions. Size of fractions should be 30% to 50% of CBV
- Elute proteins using either continuous or stepwise gradient of elution agent. The duration of continuous gradient should be 10-20 CBV. With stepwise elution apply 3-4 CBV of each concentration. Collect fractions as above.
- Clean column with 1-2 CBV of high concentration elution agent (2M NaCl for ion exchange matrix or water for hydrophobic matrix)
- Check protein concentration in each second fraction by method of Bradford. Analyse protein containing fractions using SDS-PAGE (optional)
- Combine fractions containing TP, check volume and protein concentration then calculate yield of protein after the step.
Tip: To save time on the chromatographic step do not wait until the elution is totally complete. Start to analyse protein concentration in the fractions after collection of 15-20 fractions or after about 1/3 of the gradient has been applied on the column.
Ion exchange chromatography
Please read "Ion exchange chromatography" booklet for theory and matrix characteristics.
Anion exchange matrixes and their standard columns:
Weak anion exchangers
: DEAE-Sepharose Fast Flow, DEAE-Toyopearl 650S
Strong anion exchangers
: Q-Sepharose Fast Flow, Resource Q, Mono Q.
The latter two columns are high resolution columns which have fine beads and some pressure is required to run them, therefore they are used with AKTA systems or the FPLC system and applied as a last polishing step.
Anion exchange chromatography is favoured as a first chromatographic step. Normally we use DEAE-Sepharose FF or Q-Sepharose FF columns. Crude extract prepared in buffer A (50mM tris pH 8.0) could be applied on the column directly, no extra preparation is required.
The typical pH range for anion exchange chromatography is from 6.5 to 9. To change pH in the column apply about ¼ CBV of 1M buffer solution of the desired pH, following by 2 CBV of a low concentration buffer (with the same pH as the 1M buffer solution).
The main parameters of chromatography are:
- Column size and geometry
- The columns capacity
- Flow rate
- Shape and slope of elution gradient
For standard first step anion exchange chromatography parameters are as follow:
Column Bed Volume
:
30-40ml
, diameter 1.6cm, bed height 15-20cm.
Capacity
:
up to 40 mg
of total protein per ml of matrix. Typical loading on the above column is
100-800 mg
of total protein. Higher loading affects separation.
Flow rate
: for Sepharose FF and Toyopearl 650S it is 2-3ml/min/cm
2
. So for the above column with cross-area 2cm2 typical flow rate is
4-6 ml/min
.
The best option for the first step anion exchange chromatography is a
linear elution gradient
.
For weak anion exchange matrixes (DEAE) the final NaCl concentration should be
0.5M
.
For strong anion exchange matrixes (Q) it should be
1M
.
Length of gradient is typically
10-30 CBV
.
For the first step
10 CBV
is optimal which corresponds to 300-400ml for the above column.
A gradient length of 20-30 column volumes is typically appropriate for high resolution columns for a polishing step.
Cation exchange matrixes and columns:
Weak cation exchangers: CM-Sepharose FF, CM-Toyopearl 650S
Strong cation exchangers: SP-Toyopearl 650S, S-Sepharose FF
HR ready made columns: Resource S, Mono S
Typical pH range for cation exchange chromatography is from 5 to 7.5.
All other parameters are similar to anion exchange chromatography.
Hydrophobic chromatography
Please read "Hydrophobic interaction chromatography" booklet for theory and matrixes.
I have found hydrophobic Toyopearl matrixes much better than Sepharose ones.
We use the following matrixes:
- Ether Toyopearl 650S ; weak hydrophobic matrix
- Phenyl Toyopearl 650S ; medium hydrophobic matrix
- Butyl Toyopearl 650S ; strong hydrophobic matrix
To allow proteins to bind to a hydrophobic column the sample should contain a high concentration of salt. Usually, ammonium sulphate (AS) is used. Very rarely potassium chlorate, sodium sulphate or other salts are used. The typical
concentration of ammonium sulphate in the protein sample should be 1.8-1.5M
of ammonium sulphate, which is OK for most proteins. For most hydrophilic proteins it should be increased to allow the protein to bind to the column.
For most hydrophobic proteins a 2M concentration KCl is used.
Elution gradient
is typically linear, 10-20 bed column volumes, from high salt concentration to buffer without salt or sometimes to a lower concentration of salt. Starting buffer should normally have a concentration of salt (AS) which is about 0.2M higher then the concentration of salt at the protein elution point and the final buffer should have no AS or have an AS concentration which is about 1M lower then the salt concentration at the elution point.
Capacity
of Toyopearl matrixes is up to 30 mg of total protein per ml of matrix. Optimal loading is 1-20 mg of total protein per ml of matrix.
The optimal for the lab is:
- Columns; 1x 15-20cm, about 15-20 ml CBV
- Flow rate: 1- 2 ml/min
- Loading; 20-200 mg of total protein
- Gradient; 200 ml.
NB
If protein elutes from the hydrophobic column at a low concentration of AS, you should wash column with the buffer after the completion of elution gradient in order to push protein out from the column and tubing into the collection tubes.
It is useful to
check AS concentration
in the fractions with the TP to be able to compare different runs of purifications of the same protein and so to work out optimal starting and final buffer for the gradient. Use a pocket sugar refractometer and Chart "Sugar refractometer for ammonium sulphate" (see Tables and Charts section)
Gel filtration
Please read "Gel filtration. Principles and methods for theory and matrixes" booklet.
The main feature of this type of chromatography is that it is zonal separation, so columns are long and samples are small to allow zones to be separated properly.
We use
Hi-Load Superdex 200 1.6x60cm columns
from Amersham Bioscience:
- Total volume: 120 ml
- Void volume (V0): 45ml
- Separation range MW: 600KDa - 10KDa
- Sample volume: 0.5-2ml, no restrictions on buffer composition
- Loading: up to 50 mg of total protein
- Flow rate: 1-1.5 ml/min
- Elution: with continuous buffer
Take care with this expensive column. Do not allow air to get into the column. If you, nevertheless, have dried the column, do not panic and wash column with plenty of water at flow rate 2-3 ml/min until all the air has been pushed out of it.
Standard buffer for gel filtration is 50mM tris-HCl pH 8.0, 0.5M NaCl, which is good for the majority of proteins.
Any other buffer at reasonable pH could be applied with only restriction being that low salt conditions have to be avoided to prevent absorption of proteins on the matrix. You can use this step to exchange buffer in your protein sample for the one to be used for the next purification step.
To estimate MW (molecular weight) and oligomeric state of your protein use chart "Calibration plot for Hi-Load Superdex 200 column" (see Tables and Charts section)
NB
Remember that gel filtration is not an accurate method to estimate MW or oligomeric state of the protein. Proteins are separated by their size and not by MW. Proteins with the same MW could have a different shape and compactness therefore their sizes could be different as well as elution volume (Ve) and apparent MW. Another possibility for misidentification is if the protein has some affinity to the matrix and elutes later than it should according to its MW.
Affinity chromatography
Refer to "Affinity chromatography. Principles and methods" for theory and applications.
Nowadays affinity chromatography is widely used to purify so called tagged proteins. A number of tags are used; the most common of them is 6x His tag.To fish His-taged protein from the cell free extract Ni
+2
charged imminodiacetic acid crosslinked to agarose or Sepharose beads is used. There are wide range of Ni
+2
charged matrixes and ready made columns and cartridges available from many companies, but it is at least twice as chip to buy bulk imminodiacetic acid-Sepharose from Sigma, make own columns of any size and charge them with Ni
+2
.
Pseudo affinity chromatography when matrixes with cross-linked ligands similar in structure to substrates of some enzymes are used for their purification.
In the lab we use Heparin-Sepharose Fast Flow columns (10-20ml). Capacity is about 1-1.5mg of DNA-binding protein per ml of matrix. Typically Heparin-Sepharose column is used as a first step for purification of DNA-binding proteins.
Also we can recommend so called Red and Blue columns for purification of NAD/NADP dependant proteins.
CBV
- Column Bed Volumes